Are there treatments for Alpha-1?
There are treatments to slow lung damage caused by Alpha-1.
Some people with Alpha-1 are treated with augmentation therapy. Augmentation therapy raises the levels of the alpha-1 antitrypsin (AAT) protein in the lungs using AAT protein taken from the blood of healthy donors.
People who have COPD (chronic obstructive pulmonary disease) may need other medicines or treatments, such as oxygen therapy or pulmonary rehabilitation.
Certain behavior changes, such as quitting smoking or limiting alcohol, can help prevent or delay lung damage. Talk to your doctor about steps you can take to manage Alpha-1
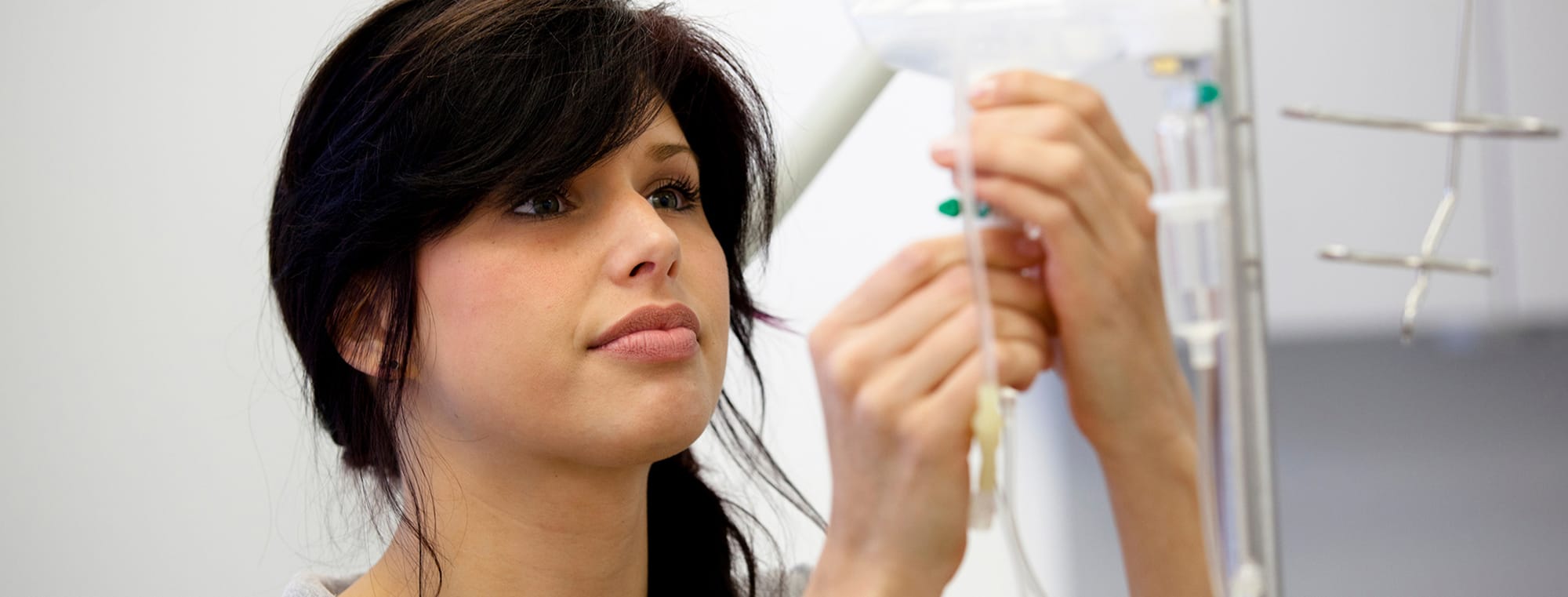
What is augmentation therapy?
Augmentation therapy is a treatment for Alpha-1 patients who have emphysema. Augmentation therapy is also known as replacement therapy.
Augmentation therapy raises the level of AAT in the lungs and blood using AAT collected from the plasma (the liquid part of blood) of healthy donors.
Boosting AAT levels in the lungs and blood can slow or stop further lung damage.
FDA-Approved Augmentation Therapies
Five augmentation therapies are approved by the U.S. Food and Drug Administration (FDA):
- Prolastin-C®
- Prolastin-C Liquid®
- Aralast NP™
- Zemaira®
- Glassia®
How is augmentation therapy given?
People treated with augmentation therapy get weekly infusions of AAT to boost AAT levels in the lungs and bloodstream.
Augmentation therapy is usually given directly into the bloodstream through an IV catheter or a port.
- An IV catheter is a thin, flexible tube inserted into a vein (typically in the lower arm or on the back of the hand) using a needle
- A port is a small device placed under the skin, usually on the right side of the chest, through a simple surgical procedure. A needle can then be inserted through the skin into the port
If You Have a Port
The benefit of a port is a new IV is not required each time an infusion is given. This protects the veins and makes the infusion process a bit faster. However, having a port increases a person’s risk of infection. If you have a port, ask your doctor about the best way to keep your port clean and avoid infections.
What is the goal of augmentation therapy?
The goal of augmentation therapy is to increase the level of AAT in your lungs to protect them from further damage.
Augmentation therapy can’t reverse the damage that has already happened or return lung function that has been lost, but it can help slow or stop more damage to the lungs.
Augmentation therapy can also be used to treat the Alpha-1-related skin disease called necrotizing panniculitis, when areas of the fatty tissue under the skin die.
Who can be treated with augmentation therapy?
Augmentation therapy is only for people with Alpha-1 who have emphysema or severe panniculitis. Ask your doctor if augmentation therapy is right for you
What are the side effects of augmentation therapy?
Most people who receive augmentation therapy have mild or no side effects.
The most common side effects of augmentation therapy are fatigue or flu-like symptoms, which typically last for up to 24 hours after an infusion. Slower infusion rates can lessen or prevent these symptoms.
Less common side effects of augmentation therapy include mild allergic reactions, such as a rash or hives, itching, tightness in the chest, altered breathing, and/or wheezing. Taking an antihistamine, such as Benadryl, before infusion can minimize the risk of allergic reaction.
Some people have side effects that are severe enough to stop augmentation therapy entirely. This is very rare.
What can be done to prepare for augmentation therapy?
There are steps people can take to prepare for augmentation therapy.
- Get tested for IgA deficiency: Testing for IgA deficiency is recommended before beginning augmentation therapy. IgA deficiency is a hereditary condition that increases a person’s risk of severe allergic reactions to plasma products, including augmentation therapy
- Get vaccinated against Hepatitis A and B: Hepatitis A and B vaccines are recommended before beginning augmentation therapy. These vaccines protect against viral infections that can cause liver damage. Hepatitis A and B vaccines are given in a series of injections




